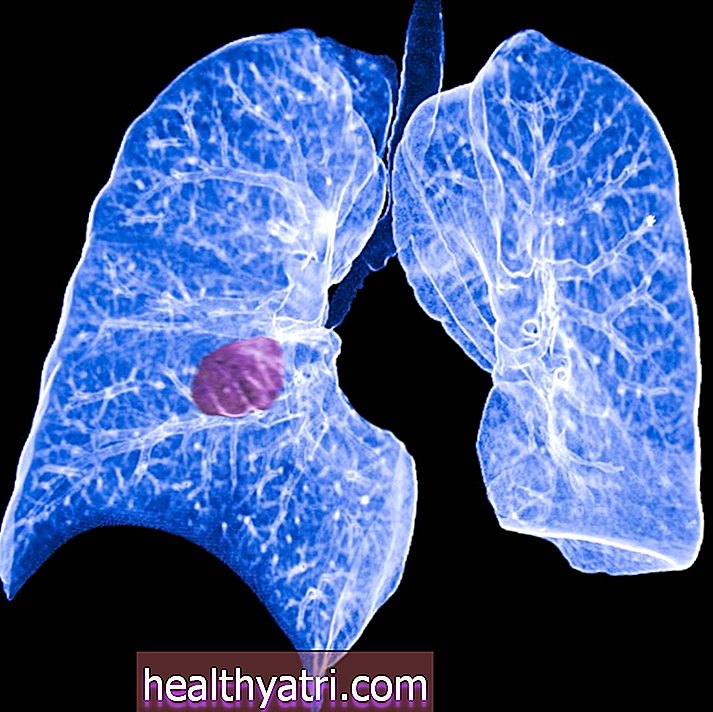
Idiopatisk lungefibrose (IPF) er en type kronisk lungesygdom, der forårsager gradvis forværret dyspnø (åndenød). Mennesker med IPF kan også opleve en tør og vedvarende hoste, progressiv træthed eller uforklarligt vægttab. Mennesker, der udvikler denne tilstand, bliver ofte handicappede på grund af åndedrætsrelaterede symptomer og vil sandsynligvis opleve tidlig død.
PASIEKA / Getty Images
IPF er ikke en almindelig sygdom, men den betragtes ikke som sjælden. Omkring 15.000 mennesker skønnes at dø af IPF hvert år i De Forenede Stater. Det påvirker mænd oftere end kvinder, rygere oftere end ikke-rygere og normalt mennesker over 50 år.
Årsagen til IPF er ikke fuldstændigt udarbejdet ("idiopatisk" betyder "af ukendt årsag"), og der er ingen kur mod det. Imidlertid udføres der en enorm mængde forskning for at forstå denne tilstand og for at udvikle effektive behandlinger for IPF. Prognosen for mennesker med IPF er allerede forbedret betydeligt i løbet af de sidste par år.
Flere nye tilgange til behandling af IPF er under udvikling, og nogle er allerede i kliniske forsøg. Det er for tidligt med sikkerhed at sige, at et gennembrud i behandlingen er lige rundt om hjørnet, men der er meget mere grund til optimisme, end der var for kort tid siden.
Vores udviklende forståelse af IPF
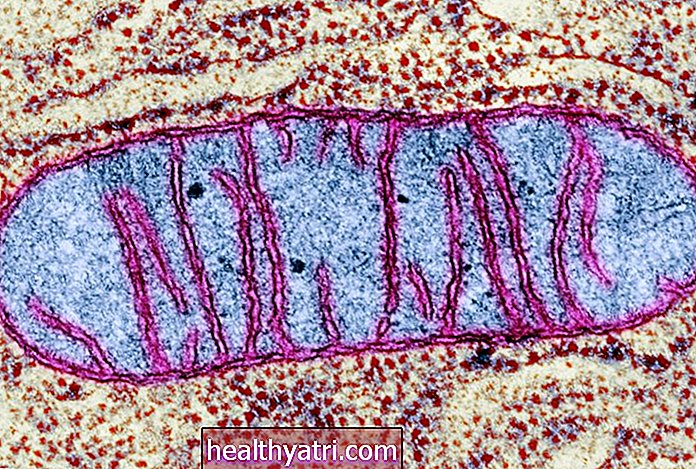
IPF er forårsaget af unormal fibrose (ardannelse) i lungevævet. I IPF erstattes de sarte celler i alveolerne (luftsække) gradvist med tykke, fibrotiske celler, der ikke er i stand til at udføre gasudveksling. Som et resultat forstyrres lungernes hovedfunktion - udveksling af gasser, der tillader ilt fra luften at komme ind i blodbanen, og kuldioxid kan forlade blodbanen -. Den gradvis forværrede evne til at få nok ilt ind i blodbanen er det, der forårsager de fleste symptomer på IPF.
I mange år var arbejdsteorien om årsagen til IPF en baseret på betændelse. Det blev antaget, at noget forårsagede betændelse i lungevævet, hvilket førte til overdreven ardannelse. Så tidlige former for behandling for IPF var i vid udstrækning rettet mod at forhindre eller bremse den inflammatoriske proces. Sådanne behandlinger har inkluderet steroider, methotrexat og cyclosporin. For det meste var disse behandlinger kun minimalt effektive (hvis overhovedet) og havde betydelige bivirkninger.
Ved at forklare årsagen til IPF har forskere i dag stort set vendt deres opmærksomhed væk fra en teoretisk betændelsesudløsende proces og mod det, der nu menes at være en proces med unormal heling af lungevæv hos mennesker med denne tilstand. Det vil sige, det primære problem, der forårsager IPF, er muligvis ikke overdreven vævsskade, men unormal heling fra (muligvis endda normal) vævsskade. Med denne unormale heling opstår overdreven fibrose, der fører til permanent lungeskade.
Den normale heling af lungevæv viser sig at være en utrolig kompleks proces, der involverer interaktion mellem forskellige typer celler og adskillige vækstfaktorer, cytokiner og andre molekyler. Den overdrevne fibrose i IPF menes nu at være relateret til en ubalance mellem disse forskellige faktorer under helingsprocessen. Faktisk er der blevet identificeret flere specifikke cytokiner og vækstfaktorer, der menes at spille vigtige roller i stimulering af overdreven lungefibrose.
Disse molekyler er nu målene for omfattende forskning, og flere lægemidler udvikles og testes i forsøget på at gendanne en mere normal helingsproces hos mennesker med IPF. Indtil videre har denne forskning ført til nogle få succeser og flere fiaskoer - men succeserne har været meget opmuntrende, og selv fejlene har avanceret vores viden om IPF.
Succeser indtil videre
I 2014 godkendte FDA to nye lægemidler til behandling af IPF, nintedanib (Ofev) og pirfenidon (Esbriet). Nintedanib formodes at fungere ved at blokere receptorer for tyrosinkinaser, molekyler der styrer forskellige vækstfaktorer for fibrose. Den nøjagtige virkningsmekanisme for pirfenidon er ikke kendt, men det menes at reducere fibrose ved at mindske fibroblastvækst og produktionen af fibrose-associerede proteiner og cytokiner og kan mindske dannelse og ophobning af ekstracellulær matrix som reaktion på vækstfaktorer.
Begge medikamenter har vist sig at nedsætte IPF-progressionen betydeligt.
Desværre kan enkeltpersoner reagere bedre på det ene eller det andet af disse to lægemidler, og på dette tidspunkt er der ingen klar måde at fortælle hvilket lægemiddel der kan være bedre for hvilken person. En lovende test kan imidlertid være i horisonten for at forudsige en persons respons på disse to stoffer. Mere om dette nedenfor.
Derudover er det nu blevet anerkendt, at mange mennesker med IPF (op til 90%) har gastroøsfageal reflukssygdom (GERD), som kan være så minimal, at de ikke bemærker det. Imidlertid kan kronisk "mikroreflux" være en faktor, der udløser mindre skader i lungevævet - og hos mennesker, der har en unormal lungehelingsproces, kan der opstå overdreven fibrose.
Små randomiserede forsøg har antydet, at personer med IPF, der behandles for GERD, kan opleve signifikant langsommere progression af deres IPF. Mens større og længerevarende kliniske forsøg er nødvendige, mener nogle eksperter, at "rutinemæssig" behandling af GERD allerede er en god idé hos mennesker, der har IPF.
Mulige fremtidige succeser
Det vides, at mange mennesker, der udvikler IPF, har en genetisk disposition for denne tilstand. Der udføres aktiv forskning for at sammenligne genetiske markører i normalt lungevæv med genetiske markører i lungevævet hos mennesker, der har IPF. Flere genetiske forskelle i IPF-vævet er allerede blevet identificeret. Disse genetiske markører giver forskere specifikke mål for lægemiddeludvikling i behandlingen af IPF. Om få år vil lægemidler, der specifikt er "skræddersyet" til behandling af IPF, sandsynligvis nå det kliniske forsøgsfase.
Mens vi venter på specifik, målrettet lægemiddelbehandling, er der i mellemtiden allerede testet nogle få lovende stoffer:
- Imatinib: Imatinib er en anden tyrosinkinasehæmmer, der ligner nintedanib.
- FG-3019: Dette lægemiddel er et monoklonalt antistof rettet mod vækstfaktor for bindevæv og er designet til at begrænse fibrose.
- Thalidomid: Dette lægemiddel har vist sig at reducere lungefibrose i dyremodeller og testes hos patienter med IPF.
- Kombineret terapi med nintedanib med pirfenidon
- PRM-151 / Pentraxin 2: Et rekombinant humant serum amyloid P / pentraxin 2 protein.
- GLPG1690: En selektiv autotaxin-hæmmer med et lille molekyle.
- Pamrevlumab: Et fuldt humant rekombinant monoklonalt antistof mod bindevævsvækstfaktor (CTGF).
Pulmosfærer
Forskere ved University of Alabama har beskrevet en ny teknik, hvor de samler "pulmosfærer" - små kugler lavet af væv fra en lunge hos en person med IPF - og udsætter pulmosfærerne for anti-IPF-lægemidlerne nintendanib og pirfenidon. ved denne test tror de, at de på forhånd kan afgøre, om patienten sandsynligvis vil reagere positivt på det ene eller begge af disse lægemidler. Hvis den tidlige erfaring med pulmosfærer bekræftes ved yderligere test, kan dette i sidste ende blive tilgængelig som en standardmetode til forudtestning af forskellige lægemiddelregimer hos mennesker med IPF.
Et ord fra Verywell
IPF er en meget alvorlig lungesygdom, og det kan være en ødelæggende at få denne diagnose. Faktisk vil en person med IPF, der foretager en Google-søgning på denne tilstand, sandsynligvis komme meget deprimeret væk. Imidlertid er der i de sidste par år gjort enorme fremskridt med hensyn til behandling af IPF. To effektive nye lægemidler er allerede godkendt til behandlingen, flere nye stoffer testes i kliniske forsøg, og målrettet forskning lover snart at give nye behandlingsmuligheder.
Hvis du eller en elsket person med IPF er interesseret i at blive overvejet til et klinisk forsøg med et af de nye lægemidler, kan oplysninger om igangværende kliniske forsøg findes på clinicaltrials.gov.




.jpg)