Chediak-Higashi syndrom er en sjælden autosomal recessiv genetisk lidelse. Det stammer fra en abnormitet i DNA'et, der forårsager abnormiteter i lysosomernes funktion eller elementer i celler, der er kritiske for mange vigtige aspekter af kroppens funktion.
Thomas Barwick / Getty ImagesImmunsystemet påvirkes især af denne sygdom, hvilket efterlader kroppen mindre i stand til at bekæmpe vira og bakterier, hvilket fører til tilbagevendende infektioner, der ofte viser sig at være dødelige i barndommen. Lysosomal dysfunktion forårsager også forskellige andre problemer, herunder neurologiske abnormiteter, albinisme og koagulationsdefekter.
Det er en meget sjælden tilstand med en forekomst på mindre end en ud af 1.000.000. Mindre end 500 tilfælde er rapporteret over hele verden.
Symptomer
Albinisme
Dem med denne genetiske abnormitet identificeres normalt i barndommen og i barndommen. Melanocytter, som er melanindannende celler, transporteres ikke passende til det sted, hvor de skal hen. (Melanin er pigmentet i øjnene, huden og håret.)
Dette får dem med Chediak-Higashi til at præsentere sig for okulokutan albinisme (oculo,der betyder "øjne" ogkutan,betyder "hud"). De fleste patienter har lys hud med tyndt lys hår, der kan være grå, hvid eller blond. Deres øjne er også normalt lyse, og de kan have fotofobi, nystagmus, strabismus eller nedsat synsstyrke.
Den "kutane" manifestation af den okulokutane albinisme kan være til stede som enten hyperpigmentering eller hypopigmentering, der er plettet.
Progressiv neurologisk dysfunktion
Neurologiske defekter, inklusive det perifere og centrale nervesystem, er progressive og forekommer hos ca. 10% til 15% af dem, der overlever i den tidlige barndom og derover. De inkluderer en bred vifte af problemer, herunder anfald, bevægelsesforstyrrelser, demens, udviklingsmæssige forsinkelse, svaghed, sensorisk underskud, rysten, ataksi og kranienerveparese.
Immunmangel
Hyppige infektioner forårsaget af specifikke bakterier, herunder staphylococcus aureus, streptococcus pyogenes og pneumococcus arter. Neutrofiler, de infektionsbekæmpende celler i vores krop, fungerer ikke korrekt i dette syndrom på grund af unormale granulater, der påvirker de hvide blodlegemeres evne til at bekæmpe infektion.
Infektionerne er normalt alvorlige og er placeret på huden, luftvejene og slimhinderne.
Infektionerne er kendt som "pyogene", hvilket betyder, at de er fyldte og normalt ildelugtende. De spænder fra at være overfladiske til dybe, hvilket kan forårsage sårdannelser. Disse efterlader dårlige ar og heler langsomt. Hvis sygdommen ikke behandles med succes, når de fleste børn den accelererede fase af sygdommen, som involverer hæmofagocytisk lymfohistiocytose (HLH), kan også forekomme, hvilket forårsager en alvorlig immundefekt. feber, forstørret milt og lever og blødning. Dette kan forekomme tidligt i barndommen eller i den tidlige barndom og er normalt dødelig.
Blodsygdomme
Patienter er ude af stand til at størkne på grund af en defekt med blodplader, hvilket fører til unormal blødning og let blå mærker.
Andre sygdomme
Andre organsystemer kan blive påvirket, såsom nyrer, mave-tarmkanalen, og periodontale sygdomme kan forekomme.
Årsager
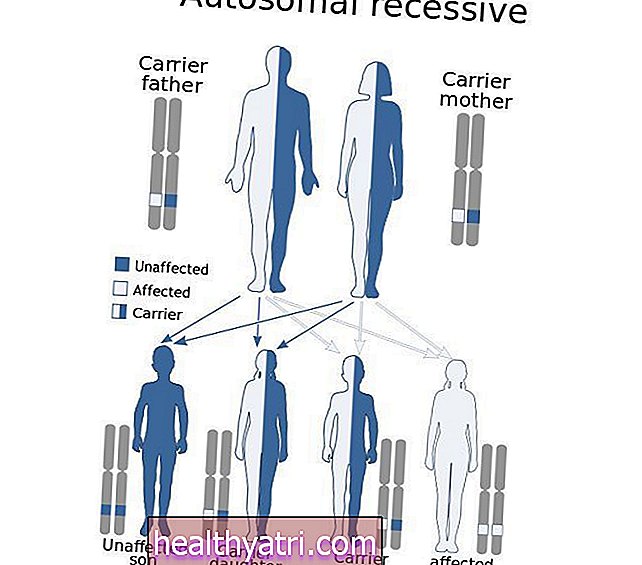
Chediak-Higashi syndrom er en sjælden autosomal recessiv genetisk lidelse forårsaget af mutationer i LYST-genet.Dette betyder, at begge forældre bærer en kopi af det muterede gen, men de viser typisk ikke tegn og symptomer på tilstanden.
LYST-genet giver instruktioner til fremstilling af et protein kendt som regulatoren for lysosomal handel. Uden denne regulator forstyrres lysosomal funktion, størrelse og struktur, og kroppen kan ikke udføre sin regelmæssige vedligeholdelse og funktioner.
Disse funktioner inkluderer bortskaffelse af uønsket indhold i celler ved hjælp af fordøjelsesenzymer til fordøjelse af bakterier, nedbrydning af giftige stoffer og genbrug af cellekomponenter. Det defekte immunsystem kan ikke beskytte kroppen mod infektioner.
Diagnose
Diagnosen Chediak-Higashi er normalt mistænkt hos patienter med delvis okulokutan albinisme og tilbagevendende pyogene infektioner. Det første skridt er at lave blodudstrygning. Dette undersøges for klassiske tegn på sygdommen, som inkluderer kæmpe azurofile granulater i neutrofiler, eosinofiler og andre granulocytter. De findes mange steder, herunder knoglemarv, melanocytter, gastrisk slimhinde, fibroblaster renalt tubulært epitel og det perifere og centrale nervevæv.
Der er flere lidelser, der ligner Chediak-Higashi. For at skelne mellem nogle af dem (inklusive Griscelli syndrom, Hermansky Pudlak syndromer) skal genetisk test udføres. Disse ser efter mutationer i CHS1 / LYST-genet.
Der er derefter diagnostiske kriterier i den accelererede fase af sygdommen, hvoraf patienten har brug for fem ud af de otte kriterier, der inkluderer feber, forstørret milt, nedsættelse af mindst to perifere blodlinjer, lav eller fraværende naturlig dræbercelleaktivitet, hyperferritinæmi og hypertriglyceridæmi og / eller hypofibrinogenæmi, hæmofagocytose i knoglemarv, milt eller lymfeknuder og høje niveauer af interleukin 2-receptor. Dette kriterium er det samme for hæmofagocytisk lymfohistiocytose.
Hvis der er en mistanke om, at et foster i livmoderen har denne sygdom på grund af en positiv familiehistorie, er det muligt at diagnosticere det prænatalt med chorionisk villusprøveudtagning, føtal blod eller hårprøveudtagning.
Behandling
Indledende behandling ved diagnose indebærer anvendelse af antibiotika profylaktisk for at forhindre bakterielle infektioner. Hvis der opstår infektioner, er aggressiv behandling berettiget. For at forhindre infektion bruges granulocytkolonistimulerende faktor (kendt som G-CSF) til at forsøge at mindske infektionen ved at øge neutrofiler, der bekæmper bakterier.
Glukokortikoider og fjernelse af milten har vist sig at være noget vellykket til at forsinke starten på den accelererede fase, og andre anvendte terapier inkluderer intravenøs gammaglobulin, antivirale midler og kemoterapi. Ingen af disse terapier er helbredende, dog.
For at korrigere den immun- og hæmatologiske virkning af Chediak-Higashi er en allogen hæmatopoietisk celletransplantation (HCT), inklusive ledningblodstransplantation, den valgte behandling. Selvom dette er vellykket, forhindrer det ikke okulokutan albinisme eller de progressive neurologiske handicap, der uundgåeligt forårsager neurologisk forringelse.
HCT menes at være mere vellykket, hvis der er opstået færre infektioner hos patienten, især HLH. Derfor er tidlig HCT ideel og kan sænke risikoen for HLH og sygdommens accelererede fase.
Succesfuldt transplanterede patienter har ingen signifikante infektioner og udvikler sig ikke til (eller har en gentagelse af) den accelererede fase.
Hvis de ikke transplanteres, dør de fleste patienter med Chediak-Higashi af pyogen infektion, før de er syv år. I en gennemgang af 35 børn med Chediak-Higashi-syndrom var den fem-årige sandsynlighed for overlevelse efter transplantation 62%.
Imidlertid udvikler de få patienter, der overlever til tidlig voksenalder, uanset om de blev transplanteret eller ej, neurologiske underskud, når de når deres tidlige tyverne.
Sørg for at diskutere med din læge, hvis du har en familiehistorie af sygdommen.
Hvad skal man gøre, når man ikke kan få en diagnose